




 View fulltext
View fulltext
Li-hui Mou, Gui-duo Jiang, Zi-yu Li, and Sheng-gui He
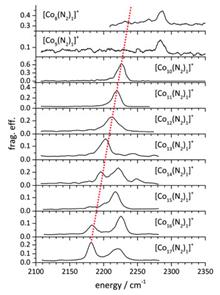
Reactions of gas-phase species with small molecules are being actively studied to understand the elementary steps and mechanistic details of related condensed-phase processes. Activation of the very inert N$\equiv$N triple bond of dinitrogen molecule by isolated gas-phase species has attracted considerable interest in the past few decades. Apart from molecular adsorption and dissociative adsorption, interesting processes such as C-N coupling and degenerate ligand exchange were discovered. The present review focuses on the recent progress on adsorption, activation, and functionalization of N2 by gas-phase species (particularly metal cluster ions) using mass spectrometry, infrared photo-dissociation spectroscopy, anion photoelectron spectroscopy, and quantum chemical calculations including density functional theory and high-level ab initio calculations. Recent advances including characterization of adsorption products, dependence of clusters' reactivity on their sizes and structures, and mechanisms of N$\equiv$N weakening and splitting have been emphasized and prospects have been discussed.
Jan. 01, 1900Vol. 33 Issue 5 507 (2020)
Bin Wu, Xu-dong Wang, Xiao-fei Gao, Hao Li, and Shan Xi Tian
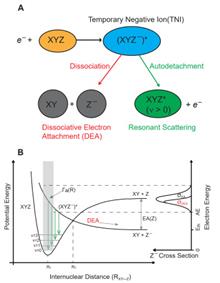
Our experimental progresses on the reaction dynamics of dissociative electron attachment (DEA) to carbon dioxide (CO2) are summarized in this review. First, we introduce some fundamentals about the DEA dynamics and provide an epitome about the DEAs to CO2. Second, the experimental technique developments are described, in particular, on the high-resolution velocity map imaging apparatus in which we put a lot of efforts during the past two years. Third, our findings about the DEA dynamics of CO2 are surveyed and briefly compared with the others' work. At last, we give a perspective about the applications of the DEA studies and highlight the inspirations in the production of molecular oxygen on Mars and the catalytic transformations of CO2.
Jan. 01, 1900Vol. 33 Issue 5 521 (2020)
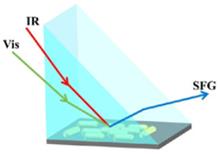
Liang Zhang, Junjun Tan, Quanbing Pei, and Shuji Ye
Sum frequency generation vibrational spectroscopy (SFG-VS) is a powerful technique for determining molecular structures at both buried interface and air surface. Distinguishing the contribution of SFG signals from buried interface and air surface is crucial to the applications in devices such as microelectronics and bio-tips. Here we demonstrate that the SFG spectra from buried interface and air surface can be differentiated by controlling the film thickness and employment of surface-plasmon enhancement. Using substrate-supported PMMA (poly(methyl methacrylate)) films as a model, we have visualized the variations in the contribution of SFG signals from buried interface and air surface. By monitoring carbonyl and C-H stretching groups, we found that SFG signals are dominated by the moieties (-CH2, -CH3, -OCH3 and C=O) segregated at the PMMA/air surface for the thin films while they are mainly contributed by the groups (-OCH3 and C=O) at the substrate/PMMA buried interface for the thick films. At the buried interface, the tilt angle of C=O decreases from 65° to 43° as the film preparation concentration increases; in contrast, the angles at the air surface fall in the range from 38° to 21°. Surface plasmon generated by gold nanorods can largely enhance SFG signals, particularly the signals from the buried interface.
Jan. 01, 1900Vol. 33 Issue 5 532 (2020)
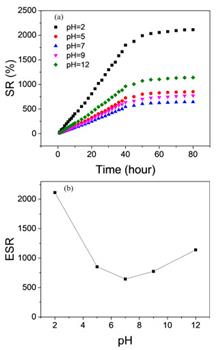
Jian Hong, De-xia Zhou, Hong-xing Hao, Min Zhao, and Hong-tao Bian
Hydrogels show versatile properties and are of great interest in the fields of bioelectronics and tissue engineering. Understanding the dynamics of the water molecules trapped in the three-dimensional polymeric networks of the hydrogels is crucial to elucidate their mechanical and swelling properties at the molecular level. In this report, the poly(DMAEMA-co-AA) hydrogels were synthesized and characterized by the macroscopic swelling measurements under different pH conditions. Furthermore, the microscopic structural dynamics of pH stimulus-responsive hydrogels were studied using FTIR and ultrafast IR spectroscopies from the viewpoint of the SCN- anionic solute as the local vibrational reporter. Ultrafast IR spectroscopic measurements showed the time constants of the vibrational population decay of SCN- were increased from 14±1 ps to 20±1 ps when the pH of the hydrogels varied from 2.0 to 12.0. Rotational anisotropy measurements further revealed that the rotation of SCN- anionic probe was restricted by the three-dimensional network formed in the hydrogels and the rotation of SCN- anionic probe cannot decay to zero especially at the pH of 7.0. These results are expected to provide a molecular-level understanding of the microscopic structure of the cross-linked polymeric network in the pH stimulus-responsive hydrogels.
Jan. 01, 1900Vol. 33 Issue 5 540 (2020)
Zhen Chi, Hui-hui Chen, Zhuo Chen, and Hai-long Chen
Defect-mediated processes in two-dimensional transition metal dichalcogenides have a significant influence on their carrier dynamics and transport properties, however, the detailed mechanisms remain poorly understood. Here, we present a comprehensive ultrafast study on defect-mediated carrier dynamics in ion exchange prepared few-layer MoS2 by femtosecond time-resolved Vis-NIR-MIR spectroscopy. The broadband photobleaching feature observed in the near-infrared transient spectrum discloses that the mid-gap defect states are widely distributed in few-layer MoS2 nanosheets. The processes of fast trapping of carriers by defect states and the following nonradiative recombination of trapped carriers are clearly revealed, demonstrating the mid-gap defect states play a significant role in the photoinduced carrier dynamics. The positive to negative crossover of the signal observed in the mid-infrared transient spectrum further uncovers some occupied shallow defect states distributed at less than 0.24 eV below the conduction band minimum. These defect states can act as effective carrier trap centers to assist the nonradiative recombination of photo-induced carriers in few-layer MoS2 on the picosecond time scale.
Jan. 01, 1900Vol. 33 Issue 5 547 (2020)
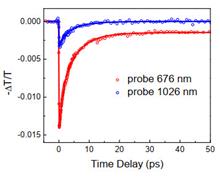
Cai-he Liu, Rui-peng Bai, Yu Bai, Yuan Guo, and Zhen Zhang
Si(111) electrode has been widely used in electrochemical and photoelectrochemical studies. The potential dependent measurements of the second harmonic generation (SHG) were performed to study Si(111) electrode interface. At different azimuthal angles of the Si(111) and under different polarization combinations, the curve of the intensity of SHG with extern potential has a different form of line or parabola. Quantitative analysis showed that these differences in the potential-dependence can be explained by the isotropic and anisotropic contribution of the Si(111) electrode. The change in the isotropic and anisotropic contribution of the Si(111) electrode may be attributed to the increase in the doping concentration of Si(111) electrodes.
Jan. 01, 1900Vol. 33 Issue 5 554 (2020)

Xiao-xia Li, Shen-long Jiang, and Qun Zhang
Two thin-film 2D organic-inorganic hybrid perovskites, i.e., 2-phenylethylammonium lead iodide (PEPI) and 4-phenyl-1-butylammonium lead iodide (PBPI) were synthesized and investigated by steady-state absorption, temperature-dependent photoluminescence, and temperature-dependent ultrafast transient absorption spectroscopy. PBPI has a longer organic chain (via introducing extra ethyl groups) than PEPI, thus its inorganic skeleton can be distorted, bringing on structural disorder. The comparative analyses of spectral profiles and temporal dynamics revealed that the greater structural disorder in PBPI results in more defect states serving as trap states to promote exciton dynamics. In addition, the fine-structuring of excitonic resonances was unveiled by temperature-dependent ultrafast spectroscopy, suggesting its correlation with inorganic skeleton rather than organic chain. Moreover, the photoexcited coherent phonons were observed in both PEPI and PBPI, pointing to a subtle impact of structural disorder on the low-frequency Raman-active vibrations of inorganic skeleton. This work provides valuable insights into the optical properties, excitonic behaviors and dynamics, as well as coherent phonon effects in 2D hybrid perovskites.
Jan. 01, 1900Vol. 33 Issue 5 561 (2020)
Shan Sun, Hui-zhong Ma, Xiao Zhang, and Yu-chen Ma
Highly luminescent bulk two-dimensional covalent organic frameworks (COFs) attract much attention recently. Origin of their luminescence and their large Stokes shift is an open question. After first-principles calculations on two kinds of COFs using the GW method and Bethe-Salpeter equation, we find that monolayer COF has a direct band gap, while bulk COF is an indirect band-gap material. The calculated optical gap and optical absorption spectrum for the direct excitons of bulk COF agree with the experiment. However, the calculated energy of the indirect exciton, in which the photoelectron and the hole locate at the conduction band minimum and the valence band maximum of bulk COF respectively, is too low compared to the fluorescence spectrum in experiment. This may exclude the possible assistance of phonons in the luminescence of bulk COF. Luminescence of bulk COF might result from exciton recombination at the defects sites. The indirect band-gap character of bulk COF originates from its AA-stacked conformation. If the conformation is changed to the AB-stacked one, the band gap of COF becomes direct which may enhance the luminescence.
Jan. 01, 1900Vol. 33 Issue 5 569 (2020)
Wei Wang, Jie Wang, Chu Gong, Chaonan Mu, Dongmei Zhang, and Xinxing Zhang
The simple homodinuclear M$ - $M single bonds for group II and XII elements are difficult to obtain as a result of the fulfilled s$ ^2 $ electronic configurations, consequently, a dicationic prototype is often utilized to design the M$ ^+ $$ - $M$ ^+ $ single bond. Existing studies generally use sterically bulky organic ligands L$ ^- $ to synthesize the compounds in the L$ ^- $$ - $M$ ^+ $$ - $M$ ^+ $$ - $L$ ^- $ manner. However, here we report the design of Mg$ - $Mg and Zn$ - $Zn single bonds in two ligandless clusters, Mg$ _2 $B$ _7 $$ ^- $ and Zn$ _2 $B$ _7 $$ ^- $, using density functional theory methods. The global minima of both of the clusters are in the form of M$ _2 $$ ^{2+} $(B$ _7 $$ ^{3-} $), where the M$ - $M single bonds are positioned above a quasi-planar hexagonal B$ _7 $ moiety. Chemical bonding analyses further confirm the existence of Mg$ - $Mg and Zn$ - $Zn single bonds in these clusters, which are driven by the unusually stable B$ _7 $$ ^{3-} $ moiety that is both $ \sigma $ and $ \pi $ aromatic. Vertical detachment energies of Mg$ _2 $B$ _7 $$ ^- $ and Zn$ _2 $B$ _7 $$ ^- $ are calculated to be 2.79 eV and 2.94 eV, respectively, for the future comparisons with experimental data.
Jan. 01, 1900Vol. 33 Issue 5 578 (2020)
Lijun Geng, Hanyu Zhang, Haiming Wu, Zhendong Sun, and Zhixun Luo
We report a study on photo-ionization of benzene and aniline with incidental subsequent dissociation by the customized reflection time-of-flight mass spectrometer utilizing a deep ultraviolet 177.3 nm laser. Highly efficient ionization of benzene is observed with a weak C$ _4 $H$ _3 $$ ^+ $ fragment formed by undergoing disproportional C$ - $C bond dissociation. In comparison, a major C$ _5 $H$ _6 $$ ^{+\cdot} $ fragment and a minor C$ _6 $H$ _6 $$ ^{+\cdot} $ radical are produced in the ionization of aniline pertaining to the removal of CNH$ ^\cdot $ and NH$ ^\cdot $ radicals, respectively. First-principles calculation is employed to reveal the photo-dissociation pathways of these two molecules having a structural difference of just an amino group. It is demonstrated that hydrogen atom transfer plays an important role in the cleavage of C$ - $C or C$ - $N bonds in benzene and aniline ions. This study is helpful to understand the underlying mechanisms of chemical bond fracture of benzene ring and related aromatic molecules.
Jan. 01, 1900Vol. 33 Issue 5 583 (2020)
Juan Ren, Xian-yi Zhang, and Xiang-lei Kong
The infrared multiphoton dissociation (IRMPD) spectrum of the protonated heterodimer of ProPheH$ ^+ $, in the range of 2700-3700 cm$ ^{-1} $, has been obtained with a Fourier-transform ion cyclotron mass spectrometer combined with an IR OPO laser. The experimental spectrum shows one peak at 3565 cm$ ^{-1} $ corresponding to the free carboxyl O-H stretching vibration, and two broad peaks centered at 2935 and 3195 cm$ ^{-1} $. Theoretical calculations were performed on the level of M062X/6-311++G(d, p). Results show that the most stable isomer is characterized by a charge-solvated structure in which the proton is bound to the unit of proline. Its predicted spectrum is in good agreement with the experimental one, although the coexistence of salt-bridged structures cannot be entirely excluded.
Jan. 01, 1900Vol. 33 Issue 5 590 (2020)
Ya-zhen Li, Jia-wei Yang, Lily Makroni, Wen-liang Wang, and Feng-yi Liu
Methyl vinyl ketone oxide, an unsaturated four-carbon Criegee intermediate produced from the ozonolysis of isoprene has been recognized to play a key role in determining the tropospheric OH concentration. It exists in four configurations ($ anti $-$ anti $, $ anti $-$ syn $, $ syn $-$ anti $, and $ syn $-$ syn $) due to two different substituents of saturated methyl and unsaturated vinyl groups. In this study, we have carried out the electronic structure calculation at the multi-configurational CASSCF and multi-state MS-CASPT2 levels, as well as the trajectory surface-hopping nonadiabatic dynamics simulation at the CASSCF level to reveal the different fates of $ syn $/$ anti $ configurations in photochemical process. Our results show that the dominant channel for the S$ _1 $-state decay is a ring closure, isomerization to dioxirane, during which, the $ syn $(C$ - $O) configuration with an intramolecular hydrogen bond shows slower nonadiabatic photoisomerization. More importantly, it has been found for the first time in photochemistry of Criegee intermediate that the cooperation of two heavy groups (methyl and vinyl) leads to an evident pyramidalization of C3 atom in methyl-vinyl Criegee intermediate, which then results in two structurally-independent minimal-energy crossing points (CIs) towards the $ syn $(C$ - $O) and $ anti $(C$ - $O) sides, respectively. The preference of surface hopping for a certain CI is responsible for the different dynamics of each configuration.
Jan. 01, 1900Vol. 33 Issue 5 595 (2020)
Bing-yang Xiao, Jia-bo Xu, and Lin-jun Wang
Inspired by the branching corrected surface hopping (BCSH) method [J. Xu and L. Wang, J. Chem. Phys. 150, 164101 (2019)], we present two new decoherence time formulas for trajectory surface hopping. Both the proposed linear and exponential formulas characterize the decoherence time as functions of the energy difference between adiabatic states and correctly capture the decoherence effect due to wave packet reflection as predicted by BCSH. The relevant parameters are trained in a series of 200 diverse models with different initial nuclear momenta, and the exact quantum solutions are utilized as references. As demonstrated in the three standard Tully models, the two new approaches exhibit significantly higher reliability than the widely used counterpart algorithm while holding the appealing efficiency, thus promising for nonadiabatic dynamics simulations of general systems.
Jan. 01, 1900Vol. 33 Issue 5 603 (2020)
Zhi-jun Zhang, Zi-fei Chen, and Jian Liu
Formaldehyde and hydrogen peroxide are two important realistic molecules in atmospheric chemistry. We implement path integral Liouville dynamics (PILD) to calculate the dipole-derivative autocorrelation function for obtaining the infrared spectrum. In comparison to exact vibrational frequencies, PILD faithfully captures most nuclear quantum effects in vibrational dynamics as temperature changes and as the isotopic substitution occurs.
Jan. 01, 1900Vol. 33 Issue 5 613 (2020)
Xiao-na Lia, Li-xue Jiang, Qing-yu Liu, Yi Ren, and Gong-ping Wei
A fundamental study on C-C coupling, that is the crucial step in the Fischer-Tropsch synthesis (FTS) process to obtain multi-carbon products, is of great importance to tailor catalysts and then guide a more promising pathway. It has been demonstrated that the coupling of CO with the metal carbide can represent the early stage in the FTS process, while the related mechanism is elusive. Herein, the reactions of the CuC$ _3 $H$ ^- $ and CuC$ _3 $$ ^- $ cluster anions with CO have been studied by using mass spectrometry and theoretical calculations. The experimental results showed that the coupling of CO with the C$ _3 $H$ ^- $ moiety of CuC$ _3 $H$ ^- $ can generate the exclusive ion product COC$ _3 $H$ ^- $. The reactivity and selectivity of this reaction of CuC$ _3 $H$ ^- $ with CO are greatly higher than that of the reaction of CuC$ _3 $$ ^- $ with CO, and this H-assisted C-C coupling process was rationalized by theoretical calculations.
Jan. 01, 1900Vol. 33 Issue 5 628 (2020)
Jing Long, Zhao Ye, Yong Du, Xu-ming Zheng, and Jia-dan Xue
Photo-induced proton coupled electron transfer (PCET) is essential in the biological, photosynthesis, catalysis and solar energy conversion processes. Recently, $ p $-nitrophenylphenol (HO-Bp-NO2) has been used as a model compound to study the photo-induced PCET mechanism by using ultrafast spectroscopy. In transient absorption spectra both singlet and triplet states were observed to exhibit PCET behavior upon laser excitation of HO-Bp-NO2. When we focused on the PCET in the triplet state, a new sharp band attracted us. This band was recorded upon excitation of HO-Bp-NO2 in aprotic polar solvents, and has not been observed for $ p $-nitrobiphenyl which is without hydroxyl substitution. In order to find out what the new band represents, acidic solutions were used as an additional proton donor considering the acidity of HO-Bp-NO2. With the help of results in strong ($ \sim $10$ ^{-1} $ mol/L) and weak ($ \sim $10$ ^{-4} $ mol/L) acidic solutions, the new band is identified as open shell singlet O-Bp-NO2H, which is generated through protonation of nitro O in $ ^3 $HO-Bp-NO2 followed by deprotonation of hydroxyl. Kinetics analysis indicates that the formation of radical $ \cdot $O-Bp-NO2 competes with O-Bp-NO2H in the way of concerted electron-proton transfer and/or proton followed electron transfers and is responsible for the low yield of O-Bp-NO2H. The results in the present work will make it clear how the $ ^3 $HO-Bp-NO2 deactivates in aprotic polar solvents and provide a solid benchmark for the deeply studying the PCET mechanism in triplets of analogous aromatic nitro compounds.
Jan. 01, 1900Vol. 33 Issue 5 635 (2020)
Jin-lu He, Yong-hao Zhu, and Run Long
Recent experiments report the rotation of FA (FA = HC[NH2]2$ ^+ $) cations significantly influence the excited-state lifetime of FAPbI3. However, the underlying mechanism remains unclear. Using ab initio nonadiabatic (NA) molecular dynamics combined with time-domain density functional simulations, we have demonstrated that reorientation of partial FA cations significantly inhibits nonradiative electron-hole recombination with respect to the pristine FAPbI3 due to the decreased NA coupling by localizing electron and hole in different positions and the suppressed atomic motions. Slow nuclear motions simultaneously increase the decoherence time, which is overcome by the reduced NA coupling, extending electron-hole recombination time scales to several nanoseconds and being about 3.9 times longer than that in pristine FAPbI3, which occurs within sub-nanosecond and agrees with experiment. Our study established the mechanism for the experimentally reported prolonged excited-state lifetime, providing a rational strategy for design of high performance of perovskite solar cells and optoelectronic devices.
Jan. 01, 1900Vol. 33 Issue 5 642 (2020)
Fang-fang Li, Yu-jie Ma, Jia-xing Liu, Guan-jun Wang, and Feng-yan Wang
The photodissociation dynamics of AlO at 193 nm is studied using time-sliced ion velocity mapping. Two dissociation channels are found through the speed and angular distributions of aluminum ions: one is one-photon dissociation of the neutral AlO to generate Al($ ^2 $P$ _ \rm{u} $)+O($ ^3 $P$ _ \rm{g} $), and the other is two-photon ionization and then dissociation of AlO$ ^+ $ to generate Al$ ^+ $($ ^1 $S$ _ \rm{g} $)+O($ ^3 $P$ _ \rm{g} $). Each dissociation channel includes the contribution of AlO in the vibrational states $ v $ = 0-2. The anisotropy parameter of the neutral dissociation channel is more dependent on the vibration state of AlO than the ion dissociation channel.
Jan. 01, 1900Vol. 33 Issue 5 649 (2020)
Ying-ying Peng, Yi-fan Liao, Wei Gan, Qing-xiao Tong, and Qun-hui Yuan
Two non-ionic hydro-fluorocarbon hybrid surfactants with and without hydroxyl groups were synthesized and compared. They exhibited good thermal stability and superior surface activity. It was observed that the hydroxyl group had a profound effect on modifying the surface tension of their solutions and the morphology of the formed micelles. This effect may be attributed to the rearranging of the alkane group from above the air/aqueous surface to below it and the disrupting of the interfacial water structure induced by the hydroxyl groups. This work provides a strategy to weaken the immiscibility between hydrocarbon and fluorocarbon chains by modifying their orientational structure at the interface, thus it is helpful for the design of surfactants with varied interfacial properties.
Jan. 01, 1900Vol. 33 Issue 5 623 (2020)
© Copyright 2018-2021 | Chinese Laser Press.
All Rights Reserved 沪ICP备15018463号-20